Multiple Kernel Learning
 Background: Support Vector Machines (SVMs) -- using a variety of string kernels -- have been successfully applied to biological sequence classification problems. While SVMs achieve high classification accuracy they lack interpretability. In many applications, it does not suffice that an algorithm just detects a biological signal in the sequence, but it should also provide means to interpret its solution in order to gain biological insight. Results: We propose novel and efficient algorithms for solving the so-called Support Vector Multiple Kernel Learning problem. The developed techniques can be used to understand the obtained support vector decision function in order to extract biologically relevant knowledge about the sequence analysis problem at hand. We apply the proposed methods to the task of acceptor splice site prediction and to the problem of recognizing alternatively spliced exons. Our algorithms compute sparse weightings of substring locations, highlighting which parts of the sequence are important for discrimination. Conclusion: The proposed method is able to deal with thousands of examples while combining hundreds of kernels within reasonable time, and reliably identifies a few statistically significant positions.
Background: Support Vector Machines (SVMs) -- using a variety of string kernels -- have been successfully applied to biological sequence classification problems. While SVMs achieve high classification accuracy they lack interpretability. In many applications, it does not suffice that an algorithm just detects a biological signal in the sequence, but it should also provide means to interpret its solution in order to gain biological insight. Results: We propose novel and efficient algorithms for solving the so-called Support Vector Multiple Kernel Learning problem. The developed techniques can be used to understand the obtained support vector decision function in order to extract biologically relevant knowledge about the sequence analysis problem at hand. We apply the proposed methods to the task of acceptor splice site prediction and to the problem of recognizing alternatively spliced exons. Our algorithms compute sparse weightings of substring locations, highlighting which parts of the sequence are important for discrimination. Conclusion: The proposed method is able to deal with thousands of examples while combining hundreds of kernels within reasonable time, and reliably identifies a few statistically significant positions.
Positional Oligomer Importance Matrices
Motivation: At the heart of many important bioinformatics problems, such as gene finding and function prediction, is the classification of biological sequences. Frequently the most accurate classifiers are obtained by training support vector machines (SVMs) with complex sequence kernels. However, a cumbersome shortcoming of SVMs is that their learned decision rules are very hard to understand for humans and cannot easily be related to biological facts. 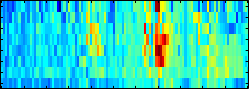 Results: To make SVM-based sequence classifiers more accessible and profitable, we introduce the concept of positional oligomer importance matrices (POIMs) and propose an efficient algorithm for their computation. In contrast to the raw SVM feature weighting, POIMs take the underlying correlation structure of k-mer features induced by overlaps of related k-mers into account. POIMs can be seen as a powerful generalization of sequence logos: they allow to capture and visualize sequence patterns that are relevant for the investigated biological phenomena. Availability: All source code, data sets, tables and figures will be made availabe at \http://www.fml.tuebingen.mpg.de/raetsch/projects/POIM.
Results: To make SVM-based sequence classifiers more accessible and profitable, we introduce the concept of positional oligomer importance matrices (POIMs) and propose an efficient algorithm for their computation. In contrast to the raw SVM feature weighting, POIMs take the underlying correlation structure of k-mer features induced by overlaps of related k-mers into account. POIMs can be seen as a powerful generalization of sequence logos: they allow to capture and visualize sequence patterns that are relevant for the investigated biological phenomena. Availability: All source code, data sets, tables and figures will be made availabe at \http://www.fml.tuebingen.mpg.de/raetsch/projects/POIM.
Feature Importance Ranking Measure
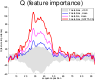 Most accurate predictions are typically obtained by learning machines with complex feature spaces (e.g., as induced by kernels). Unfortunately, such decision rules are hardly accessible to humans and cannot easily be used to gain insights about the application domain. Therefore, one often resorts to linear models in combination with variable selection, thereby sacrificing some predictive power for presump- tive interpretability. Here, we introduce the Feature Importance Ranking Measure (FIRM), which by retrospective analysis of arbitrary learning machines allows to achieve both excellent predictive performance and superior interpretation. In contrast to standard raw feature weighting, FIRM takes the underlying correlation structure of the features into account. Thereby, it is able to discover the most relevant features, even if their appearance in the training data is entirely prevented by noise. The desirable properties of FIRM are investigated analytically and illustrated in a few simulations.
Most accurate predictions are typically obtained by learning machines with complex feature spaces (e.g., as induced by kernels). Unfortunately, such decision rules are hardly accessible to humans and cannot easily be used to gain insights about the application domain. Therefore, one often resorts to linear models in combination with variable selection, thereby sacrificing some predictive power for presump- tive interpretability. Here, we introduce the Feature Importance Ranking Measure (FIRM), which by retrospective analysis of arbitrary learning machines allows to achieve both excellent predictive performance and superior interpretation. In contrast to standard raw feature weighting, FIRM takes the underlying correlation structure of the features into account. Thereby, it is able to discover the most relevant features, even if their appearance in the training data is entirely prevented by noise. The desirable properties of FIRM are investigated analytically and illustrated in a few simulations.
