ASP - Accurate Splice Site Predictor
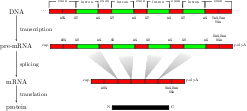 For splice site recognition, one has to solve two
classification problems: discriminating true from decoy splice
sites for both acceptor and donor sites. Gene finding systems
typically rely on Markov Chains to solve these tasks. In this
work we consider Support Vector Machines for splice site
recognition. We employ the so-called weighted degree kernel
which turns out well suited for this task, as we will
illustrate in several experiments where we compare its
prediction accuracy with that of recently proposed systems. We
apply our method to the genome-wide recognition of splice sites
in Caenorhabditis elegans, Drosophila melanogaster, Arabidopsis
thaliana, Danio rerio, and Homo sapiens. Our performance
estimates indicate that splice sites can be recognized very
accurately in these genomes and that our method outperforms
many other methods including Markov Chains, GeneSplicer and
SpliceMachine.
For splice site recognition, one has to solve two
classification problems: discriminating true from decoy splice
sites for both acceptor and donor sites. Gene finding systems
typically rely on Markov Chains to solve these tasks. In this
work we consider Support Vector Machines for splice site
recognition. We employ the so-called weighted degree kernel
which turns out well suited for this task, as we will
illustrate in several experiments where we compare its
prediction accuracy with that of recently proposed systems. We
apply our method to the genome-wide recognition of splice sites
in Caenorhabditis elegans, Drosophila melanogaster, Arabidopsis
thaliana, Danio rerio, and Homo sapiens. Our performance
estimates indicate that splice sites can be recognized very
accurately in these genomes and that our method outperforms
many other methods including Markov Chains, GeneSplicer and
SpliceMachine.
Software available from here.
ARTS - Accurate Transcription Start Site Prediction
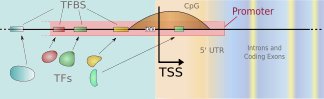 We develop new methods for finding transcription start sites (TSS) of
RNA Polymerase II binding genes in genomic DNA sequences.
Employing Support Vector Machines with advanced sequence
kernels, we achieve drastically higher prediction accuracies than
state-of-the-art methods.
We develop new methods for finding transcription start sites (TSS) of
RNA Polymerase II binding genes in genomic DNA sequences.
Employing Support Vector Machines with advanced sequence
kernels, we achieve drastically higher prediction accuracies than
state-of-the-art methods.
Software available from here.
POIMs - Positional Oligomer Importance Matrices
SVMs find a discrimination in a high dimensional kernel feature space
and as such often have to be treated as a black box. This implies that
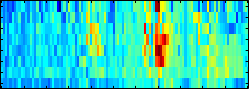 analyses
or visualization of the learning result is inherently
difficult. It poses a problem for applications in
bioinformatics as it is often very important to understand
which features are used for learning and why the accuracy is
high. We have developed the concept of Positional Oligomer Importance Matrices
(POIMs) ---
that allows us to pin-point and visualize the most discriminative motifs. Computing
poims is very efficient and can be directly applied to the learned SVM classifier.
analyses
or visualization of the learning result is inherently
difficult. It poses a problem for applications in
bioinformatics as it is often very important to understand
which features are used for learning and why the accuracy is
high. We have developed the concept of Positional Oligomer Importance Matrices
(POIMs) ---
that allows us to pin-point and visualize the most discriminative motifs. Computing
poims is very efficient and can be directly applied to the learned SVM classifier.
mSplicer - Splice Form Prediction
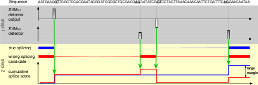 For modern biology, precise genome annotations are of prime importance as they allow the accurate definition of genic regions. We employ state of the art machine learning methods to assay and improve the accuracy of the genome annotation of the nematode Caenorhabditis elegans. The proposed machine learning system is trained to recognize exons and introns on the unspliced mRNA utilizing recent advances in support vector machines and label sequence learning.
For modern biology, precise genome annotations are of prime importance as they allow the accurate definition of genic regions. We employ state of the art machine learning methods to assay and improve the accuracy of the genome annotation of the nematode Caenorhabditis elegans. The proposed machine learning system is trained to recognize exons and introns on the unspliced mRNA utilizing recent advances in support vector machines and label sequence learning.
This software is available from msplicer.org and its mloss.org project page.
mGene - Accurate Genefinding
 mGene is a gene finding system that was developed at the
Friedrich Miescher Lab in Tübingen. It tackles the gene
prediction problem using a two-layered approach. In a first
step (layer 1) state-of-the-art kernel machines are employed to
detect signal sequences in genomic DNA. In a second step (layer
2) their outputs are combined by a Hidden Semi Markov machine
learning algorithm, to predict whole gene structures. Major
algorithms, which were used, are implemented in the SHOGUN toolbox and
combined and complemented with Octave/Matlab scripts.
mGene is a gene finding system that was developed at the
Friedrich Miescher Lab in Tübingen. It tackles the gene
prediction problem using a two-layered approach. In a first
step (layer 1) state-of-the-art kernel machines are employed to
detect signal sequences in genomic DNA. In a second step (layer
2) their outputs are combined by a Hidden Semi Markov machine
learning algorithm, to predict whole gene structures. Major
algorithms, which were used, are implemented in the SHOGUN toolbox and
combined and complemented with Octave/Matlab scripts.
mGene is described at mgene.org and web-interface that lets practitioners apply and re-train mGene is available via Galaxy.
