Accurate Splice Site Recognition
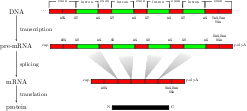 Background: For splice site recognition, one has to solve two classification problems: discriminating true from decoy splice sites for both acceptor and donor sites. Gene finding systems typically rely on Markov Chains to solve these tasks. Results: In this work we consider Support Vector Machines for splice site recognition. We employ the so-called weighted degree kernel which turns out well suited for this task, as we will illustrate in several experiments where we compare its prediction accuracy with that of recently proposed systems. We apply our method to the genome-wide recognition of splice sites in Caenorhabditis elegans, Drosophila melanogaster, Arabidopsis thaliana, Danio rerio, and Homo sapiens. Our performance estimates indicate that splice sites can be recognized very accurately in these genomes and that our method outperforms many other methods including Markov Chains, GeneSplicer and SpliceMachine. We provide genome-wide predictions of splice sites and a stand-alone prediction tool ready to be used for incorporation in a gene finder.
Background: For splice site recognition, one has to solve two classification problems: discriminating true from decoy splice sites for both acceptor and donor sites. Gene finding systems typically rely on Markov Chains to solve these tasks. Results: In this work we consider Support Vector Machines for splice site recognition. We employ the so-called weighted degree kernel which turns out well suited for this task, as we will illustrate in several experiments where we compare its prediction accuracy with that of recently proposed systems. We apply our method to the genome-wide recognition of splice sites in Caenorhabditis elegans, Drosophila melanogaster, Arabidopsis thaliana, Danio rerio, and Homo sapiens. Our performance estimates indicate that splice sites can be recognized very accurately in these genomes and that our method outperforms many other methods including Markov Chains, GeneSplicer and SpliceMachine. We provide genome-wide predictions of splice sites and a stand-alone prediction tool ready to be used for incorporation in a gene finder.
Accurate Recognition of Transcription Starts
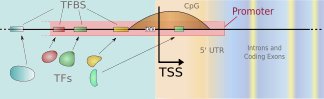 We develop new methods for finding transcription start sites (TSS) of RNA Polymerase II binding genes in genomic DNA sequences. Employing Support Vector Machines with advanced sequence kernels, we achieve drastically higher prediction accuracies than state-of-the-art methods. Motivation: One of the most important features of genomic DNA are the protein-coding genes. While it is of great value to identify those genes and the encoded proteins, it is also crucial to understand how their transcription is regulated. To this end one has to identify the corresponding promoters and the contained transcription factor binding sites. TSS finders can be used to locate potential promoters. They may also be used in combination with other signal and content detectors to resolve entire gene structures. Results: We have developed a novel kernel based method – called ARTS – that accurately recognizes transcription start sites in human. The application of otherwise too computationally expensive Support Vector Machines was made possible due to the use of efficient training and evaluation techniques using suffix tries. In a carefully designed experimental study, we compare our TSS finder to state-of-the-art methods from the literature: McPromoter, Eponine and FirstEF. For given false positive rates within a reasonable range, we consistently achieve considerably higher true positive rates. For instance, ARTS finds about 35\ true positives at a false positive rate of 1/1000, where the other methods find about a half.
We develop new methods for finding transcription start sites (TSS) of RNA Polymerase II binding genes in genomic DNA sequences. Employing Support Vector Machines with advanced sequence kernels, we achieve drastically higher prediction accuracies than state-of-the-art methods. Motivation: One of the most important features of genomic DNA are the protein-coding genes. While it is of great value to identify those genes and the encoded proteins, it is also crucial to understand how their transcription is regulated. To this end one has to identify the corresponding promoters and the contained transcription factor binding sites. TSS finders can be used to locate potential promoters. They may also be used in combination with other signal and content detectors to resolve entire gene structures. Results: We have developed a novel kernel based method – called ARTS – that accurately recognizes transcription start sites in human. The application of otherwise too computationally expensive Support Vector Machines was made possible due to the use of efficient training and evaluation techniques using suffix tries. In a carefully designed experimental study, we compare our TSS finder to state-of-the-art methods from the literature: McPromoter, Eponine and FirstEF. For given false positive rates within a reasonable range, we consistently achieve considerably higher true positive rates. For instance, ARTS finds about 35\ true positives at a false positive rate of 1/1000, where the other methods find about a half.
mSplicer
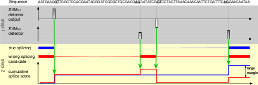 For modern biology, precise genome annotations are of prime importance as they allow the accurate definition of genic regions. We employ state of the art machine learning methods to assay and improve the accuracy of the genome annotation of the nematode Caenorhabditis elegans. The proposed machine learning system is trained to recognize exons and introns on the unspliced mRNA utilizing recent advances in support vector machines and label sequence learning.
For modern biology, precise genome annotations are of prime importance as they allow the accurate definition of genic regions. We employ state of the art machine learning methods to assay and improve the accuracy of the genome annotation of the nematode Caenorhabditis elegans. The proposed machine learning system is trained to recognize exons and introns on the unspliced mRNA utilizing recent advances in support vector machines and label sequence learning.
mGene
 We present a highly accurate gene prediction system for eukaryotic genomes, called mGene. It combines in an unprecedented manner the flexibility of generalized hidden Markov models with the predictive power of modern machine learning meth- ods, such as Support Vector Machines (SVMs). Its excellent performance was proved in an objective competition based on the genome of the nematode Caenorhabditis elegans (Coghlan et al., 2008). Considering the average of sensitivity and specificity the developmental version of mGene exhibited the best prediction performance on nucleotide, exon, and transcript level for ab initio and multiple-genome gene pre- diction tasks. The fully developed version shows superior performance in ten out of twelve evaluation criteria compared to the other participating gene finders, including Fgenesh++ (Salamov and Solovyev, 2000) and Augustus (Stanke et al., 2006). An in-depth analysis of mGene's genome-wide predictions revealed that $\ 2, 200 predicted genes were not contained in the current genome annotation. Testing a subset of 57 of these genes by RT-PCR and sequencing, we confirmed expression for 24 (42\ of them. mGene missed 300 annotated genes, out of which 205 were unconfirmed. RT-PCR testing of 24 of these genes resulted in a success rate of merely 8\ These findings suggest that even the gene catalog of a well-studied organism such as C. elegans can be substantially improved by mGene's predictions. We also provide gene predictions for the four nematodes C. briggsae, C. brenneri, C. japonica and C. remanei (Stein et al., 2003; Sternberg et al., 2003). Comparing the resulting proteomes among these organisms and to the known protein universe, we identified many species-specific gene inventions. In a quality assessment of several available annotations for these genomes, we find that mGene's predictions are most accurate. Availability: mGene is available as source code under Gnu Public License from the project website http://mgene.org and as a Galaxy-based webserver at http://mgene.org/web. Moreover, the gene predictions have been included in the Worm- base annotation available at http://wormbase.org and the project website.
We present a highly accurate gene prediction system for eukaryotic genomes, called mGene. It combines in an unprecedented manner the flexibility of generalized hidden Markov models with the predictive power of modern machine learning meth- ods, such as Support Vector Machines (SVMs). Its excellent performance was proved in an objective competition based on the genome of the nematode Caenorhabditis elegans (Coghlan et al., 2008). Considering the average of sensitivity and specificity the developmental version of mGene exhibited the best prediction performance on nucleotide, exon, and transcript level for ab initio and multiple-genome gene pre- diction tasks. The fully developed version shows superior performance in ten out of twelve evaluation criteria compared to the other participating gene finders, including Fgenesh++ (Salamov and Solovyev, 2000) and Augustus (Stanke et al., 2006). An in-depth analysis of mGene's genome-wide predictions revealed that $\ 2, 200 predicted genes were not contained in the current genome annotation. Testing a subset of 57 of these genes by RT-PCR and sequencing, we confirmed expression for 24 (42\ of them. mGene missed 300 annotated genes, out of which 205 were unconfirmed. RT-PCR testing of 24 of these genes resulted in a success rate of merely 8\ These findings suggest that even the gene catalog of a well-studied organism such as C. elegans can be substantially improved by mGene's predictions. We also provide gene predictions for the four nematodes C. briggsae, C. brenneri, C. japonica and C. remanei (Stein et al., 2003; Sternberg et al., 2003). Comparing the resulting proteomes among these organisms and to the known protein universe, we identified many species-specific gene inventions. In a quality assessment of several available annotations for these genomes, we find that mGene's predictions are most accurate. Availability: mGene is available as source code under Gnu Public License from the project website http://mgene.org and as a Galaxy-based webserver at http://mgene.org/web. Moreover, the gene predictions have been included in the Worm- base annotation available at http://wormbase.org and the project website.
